Light induced dynamics and control of correlated quantum systems
SFB 925Light induced dynamics and control of correlated quantum systems
Photo: UHH/Denstorf
1 January 2017
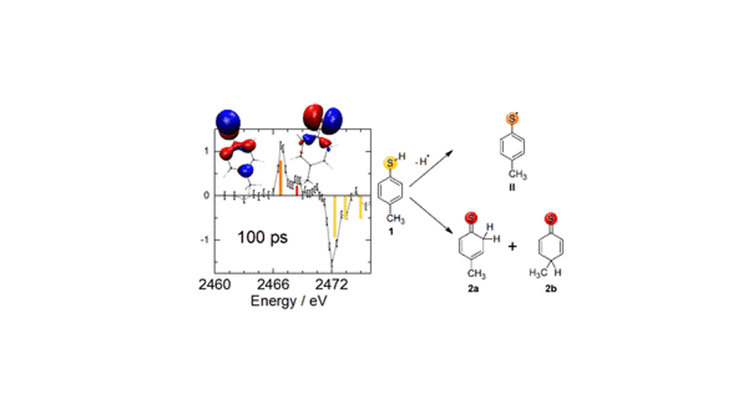
Photo: Grafik
We made the first step toward understanding the transient electronic structure during light-induced reaction dynamics of sulfur-containing molecules in solution. The high electronic variability of sulfur allows for many oxidation states and often leads to multiple quantum pathways and great complexity in the outcome of material transformations involving sulfur. We employed time-resolved sulfur-1s absorption spectroscopy as a new element-specific electronic structure probe of transient states of matter to study a model aromatic thiol system. Our study unequivo-cally identifies the transient photoproducts by their unique sulfur-1s absorption signature as the accompanying theory accurately predicts. Moreover, our study explains the so-called regioselectivity of thione isomerization in terms of the resulting radical frontier orbitals.
Reaction dynamics are governed by the underlying electronic correlations and the temporal fluctuations of the environment. Their understanding is therefore of major interest in all fields of materials science and chemistry. Open-shell systems such as radicals and their formation pathways are very important as they are highly reactive species, among which, sulfur radicals are of particular interest because they are less reactive and more electronically ‘flexible’ than their isoelectronic oxygen counterparts. Sulfur appears in many materials, ranging from polymers, nanoparticles, and electrode materials in batteries to molecular electronic devices. In nature, sulfur serves as a biochemically relevant structure and function-forming element in proteins and catalytic reaction centers as well as a radical scavenger to protect a cell’s elaborate machinery from damage by more reactive radical species.
The abundant body of literature on studies employing static sulfur-1s excitations (forming the sulfur K-edge) underscores the usefulness and important information content of sulfur K-edge spectroscopy. Optical probes tend to be very difficult to interpret due to their often broad and nondescript nature. X-ray spectroscopy mitigates this inherent dilemma by using transitions involving highly localized core-shell electrons. The sensitivity of X-ray absorption spectra to bond order, symmetry, and valence charge distribution around the absorbing atomic species can be particularly useful in the identification of different reaction intermediates and products when multiple reaction pathways are present.
To establish element-specific transient sulfur probes in the tender X-ray regime capable of femtosecond time resolution we started with a simple model system – an aromatic thiol – and combined the experimental results with restricted active space self-consistent field (RASSCF) calculations. The initial response of the thiol 4-MTP to excitation with a 267-nm femtosecond pulse is the cleavage of the S-H bond: From the initially excited state a conical intersection permits populating a dissociative excited state which leads to the ejection of the sulfur-bound hydrogen atom. Oliver et al. studied this system with femtosecond visible spectroscopy, identifying the 4-methylthiophenoxy radical (II) and an additional previously uncharacterized adduct.1
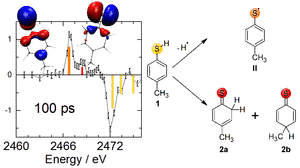
Figure 1 shows the transient X-ray absorption spectrum of the new molecular species (positive absorption change) and the missing ground state spectrum (negative absorption change) 100 ps after photoexcitation. The sulfur 1s→3p transitions predicted from RASSCF calculations are shown in color-coded bars, according to the different sulfur species.
Fig. 1: Differential absorption spectrum 100 ps after ultraviolet excitation of 4-MTP along with the deduced reaction scheme and the frontier orbitals of the transient photoproduct states. Negative signals indicate the loss of 4-MTP (yellow lines). Two new transitions appear that are unique to the 4-MTP radical (orange) and the para- and ortho-thiones (red).
4-MTP (1) and two of the many possible photoproducts coincide with the experimental difference spectrum. The 4-MTP radical (II) exhibits a very simple S-1s spectrum with one dominant transition that matches the experimentally observed induced absorption peak at 2467 eV. The second predicted transition that fits our experimental data is the observed induced absorption peak at 2468.5 eV. It corresponds to the “uncharacterized adduct” suggested by Oliver et al. as the two regioselectively formed thione isomers, denoted as ortho-(2a) and para- (2b) thione, respectively. These are formed upon recombination of the 4-MTP radical (II) with a previously ejected hydrogen atom.
The hydrogen atom greatly disfavors reattaching itself to the carbon atoms neighboring the methyl carbon (‘at the ‘bottom’ of the aromatic ring). This regioselectivity of hydrogen re-attachment is governed by the frontier orbital of the radical (II) which is a singly-occupied molecular orbital (SOMO): It exhibits electron density only at particular carbon atoms as can be seen from the SOMOS’s isosurface shown on the very left of Figure 1. Upon Hydrogen reattachment, the two thione isomers (2a & 2b) form, exhibiting nearly identical transition energies. For both thione isomers, the dominating feature is the C=S double bond. The position of the newly formed sp3-hybridized carbon does not influence the electronic structure of the sulfur atom substantially because the sulfur atom and the newly formed CH2 (2a) or CHCH3 (2b) group are separated by either two (2a) or four (2b) chemical bonds, respectively. Accordingly, the isosurface of the highest occupied molecular orbital (HOMO) of the ortho-thione (2a) appears symmetric (center top of Fig. 1) despite lack of such symmetry.
In summary, we have successfully taken the first steps in following reaction dynamics in liquid phase using sulfur-1s absorption spectroscopy, assigning the two prominent spectral signatures to the 4-MTP radical and the thione isomers. This type of spectroscopy benefits from element specificity and good theoretical predictions of the resonant transitions. The high spectral sensitivity to different oxidation states and chemical surroundings renders this technique a valuable tool to study molecules and materials involving sulfur. Sulfur K-edge spectroscopy can be applied to solution and solids, including device applications due to penetration depths of the X-ray photons up to tens of microns. This could be especially useful in the field of battery research and X-ray radiation damage of proteins (in operando, in crystallo). Free-electron lasers will permit access to atomic and possibly electronic time scales using ultrashort X-ray pulses, allowing for observation of initial reaction steps and the emergence of different photoproducts in time.
References
1. T. A. A. Oliver, Y. Zhang, M. N. R. Ashfold, and S. E. Bradforth, Faraday Discuss. 150, 439 (2011).
Miguel Ochmann, Institute for Nanostructure and Solid State Physics, Project A4
Miguel Ochmann, Institute for Nanostructure and Solid State Physics, Project A4
Original publication: „Light-Induced Radical Formation and Isomerization of an Aromatic Thiol in Solution Followed by Time-Resolved X-ray Absorption Spectroscopy at the Sulfur K-Edge”, J. Am. Chem. Soc. 139, 4797 (2017). DOI: 10.1021/jacs.6b12992
Authors: M. Ochmann, I. von Ahnen, A. A. Cordones, A. Hussain, J. H. Lee, K. Hong, K. Adamczyk, O. Vendrell, T. K. Kim, R. W. Schoenlein, N. Huse