Junior Research Group - Thorn
The Thorn Lab researches macromolecular structures, e.g. from SARS and SARS-CoV-2, with the goal to develop new methods of structure evaluation and refinement. With this, we are aiming to improve the available data for scientists in various fields of research. As a team of structural biology method developers as well as expert users, we seek to get every last bit of information from the data.
For more information and up-to-date news on our work, visit us at thorn-lab.com and insidecorona.net!
Methods and Case Studies in Structural Biology
The determination of macromolecular structures by crystallography and Cryo-EM forms the basis of today's molecular biology and biochemistry. However, these structures cannot be obtained from experimental data alone—a model is required to interpret the measured images, and hence, understanding the underlying principles is crucial. Obtaining this understanding is the driving force behind our work, as it will allow us to improve all known structures and solve more challenging ones in the future.
We employ any means necessary to solve a problem, from lab work to software development. Most of our research is driven by practical challenges and often collaborative in nature. These are our current main research topics:
Artificial Intelligence in Structural Biology
We are utilizing AI (or, more exactly, machine learning) methods in order to ais structural biology. We use them to support experimental data measurements and processing in the large collaborative project AUSPEX, to interprete reconstruction maps in Cryo-EM and research new ways to use AI-based fold prediction. So far, this included the interpretation of 2D and 3D data with convolutional neural networks and statistical methods as well as clustering. But we also look to use GANs to simulate experimental data, and after the Coronavirus Strcutural Task Force ended in February, started to dig into questions relating to linguistic models in integrative strcutural biology.
Better Models for the Crystals of Biological Macromolecules
In crystallography, the R-value reports on how well a model agrees with the experimental data. In small molecule crystallography, R-values of 3% are routinely reached. However, for biological macromolecules, R-values are usually around 20%. Something is amiss; our current models of macromolecular crystal structures are incomplete, partially incorrect, or the data have errors that we are not accounting for. These shortcomings particularly impede the determination of challenging structures, such as membrane proteins, where often only low resolution data are available. In these cases, the poor phase estimates currently obtainable result in noisy electron density maps that are difficult or even impossible to interpret. In collaboration with Armin Wagner's group at Diamond Lightsource, we try to shed light on these problems and improve our molecular models of biological crystals, so that we cannot only solve borderline cases but also improve all known structures.
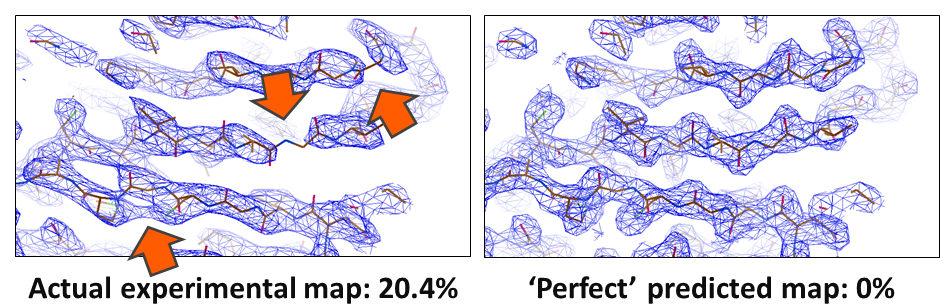
Left: Crystallographic map (blue mesh); poor phases dominate the map and it has several problems (arrows) that make it difficult to interpret. The model (sticks) is shown for clarity. Right: An ideal map would be much easier to interpret.
Modelling Large Complexes in Cryo Electron Microscopy
In Cryo-EM, high resolution of up to 2.2 Å data became available only very recently, and these advances led to the 2017 Nobel Prize in Chemistry. Cryo-EM can solve structures from 100 kDa up to several MDa, requires only a small amount of sample, and does not depend on crystallization. As cryo-EM maps have a higher information content than X-ray data, they should, in principle, be superior to electron density maps of comparable resolution. However, atomic models are currently fitted to reconstruction maps using restraints and parameters originally developed for crystallography, which limits the answers we can obtain from the new high resolution data as the underlying assumptions are not always justified. We develop tools directly based on the nature of the cryo-EM experiment, such as the neural network Haruspex, to overcome these challenges.
Improving Data Quality for Macromolecular Crystallography
Currently, less than 1% of measurement time at synchrotron sources results in a published structure; much of the other collected data suffers from avoidable quality issues—e.g., defective crystalline samples, flawed diffraction experiments, or incorrect interpretation of the images by automatic data processing procedures. If such problems were diagnosed earlier and more reliably, the current beamlines would be significantly more efficient. The lack of suitable diagnostics is one of the major roadblocks to increasing the productivity of macromolecular crystallography beamlines at synchrotrons, X-ray free-electron laser (XFEL) and neutron sources as well as for the research quality in macromolecular crystallography as a whole. Any shortcomings on part of the data have immediate negative consequences for the resulting structure and thus for the biological insights obtainable. In collaboration with beamline staff at the European X-Ray Free-Electron Laser Facility (European XFEL), the synchrotron sources BESSY and ESRF as well as the European Spallation Source ESS, we develop AUSPEX, an innovative diagnostic tool that will allow beamline scientists and users to recognize errors as early as possible, ideally before the main data collection starts. We also use these new tools to define new best practices and to improve data processing.
For more information on this, please visit www.auspex.de.
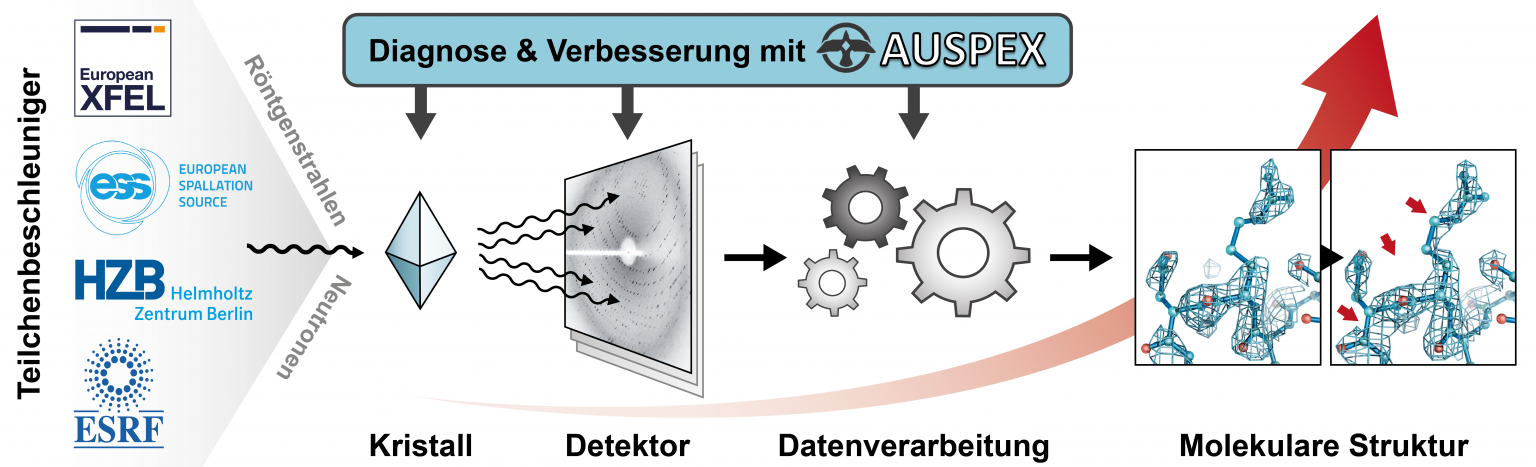
Working principle of the AUSPEX diagnostic tool.